
肿瘤糖代谢异常是肿瘤代谢重编程(metabolic reprogramming in cancer)的重要组成部分。肿瘤代谢重编程是由于肿瘤细胞内一些基因结构与功能改变导致以Warburg效应为主要生化代谢特征的一系列代谢改变,主要表现为糖酵解增强、葡萄糖摄取和消耗增加、脂类和蛋白质合成加强,以及谷氨酰胺等氨基酸摄取和分解代谢增加等,这些代谢改变将利于肿瘤恶性增殖和适应不利生存环境(图3-2-3)。
肿瘤Warburg效应 20世纪初德国生化专家Otto H. Warburg发现肝癌细胞在氧气充足情况下始终优先通过糖酵解代谢葡萄糖生成ATP。肿瘤细胞这种特殊生化表型,称为Warburg效应,或有氧糖酵解,或反Pasteur效应。当时,Warburg等将肿瘤细胞糖酵解代谢活跃的原因归结于肿瘤线粒体呼吸功能的损伤,并认为糖酵解代谢增强是致癌因素,即将肿瘤的发生归因为“生化代谢疾病”。后来随着分子生物学技术的兴起,以及“肿瘤是基因疾病”的观念在学术界得到普遍认可,肿瘤糖酵解的研究逐入低谷。1988 年,由于正电子发射断层成像(positron-emission tomography,PET)技术在临床肿瘤诊断上的成功应用,肿瘤细胞糖代谢这一特殊表型再度引起人们的高度关注。目前,Warburg效应作为肿瘤的能量代谢标志,已在多种类型的细胞中得到证实。肿瘤细胞糖酵解能力是正常细胞的20~30 倍,糖酵解的增强与肿瘤的生长速度成正比,还与肿瘤的侵袭性生长密切相关。同时肿瘤细胞糖代谢的另一条通路——磷酸戊糖通路代谢活性也大大增强,该通路为肿瘤合成代谢提供大量构件分子和还原当量(图3-2-3)。肿瘤细胞这两条葡萄糖代谢通路大量消耗了葡萄糖,这也是肿瘤恶性发展和肿瘤恶液质发生机制的重要因素。
Warburg 效应有利于肿瘤的恶性生长。尽管糖酵解较线粒体氧化磷酸化产能效率较低,但恶性肿瘤细胞可从活跃的糖酵解代谢中受益,主要表现在以下几个方面:第一,与氧化磷酸化相比,糖酵解产生ATP 效率尽管低,但产生速度快,这对于快速增殖的肿瘤细胞极为有利,因为肿瘤细胞对氧的依赖性降低了,而对于依赖氧化磷酸化作用产生AT P的细胞来说,氧的缺乏可能是致命的。肿瘤细胞受局部缺氧等内外因素影响,线粒体氧化磷酸化过程受到不同程度的抑制,糖酵解代谢可尽快补充ATP的生成。第二,肿瘤细胞可通过糖酵解获取中间代谢产物,用于合成脂肪、蛋白质和核酸,以满足其活跃的合成代谢需求。譬如,累积的糖酵解代谢产物丙酮酸可用于脂类合成,而脂类对维持膜的完整性和脂依赖型蛋白转录后修饰是必不可少的。第三,糖酵解通过影响线粒体外膜通透性使肿瘤细胞获得拮抗细胞凋亡的能力,可导致恶性肿瘤对放化疗等促凋亡作用耐受。第四,糖酵解产生大量乳酸,导致微环境酸化,有助于肿瘤侵袭和免疫逃逸。早期酸化的微环境对肿瘤细胞生存不利,而肿瘤发生发展是一个不断变异选择的过程,当耐酸的肿瘤细胞株形成后,这种微环境则对肿瘤细胞有着保护作用,因为酸性环境对正常细胞具有一定毒性,可导致细胞基质的分解和外源性碱性抗癌药物的失效,从而有利于肿瘤细胞的生长与转移。第五,糖酵解还直接促进缺氧诱导因子1(hypoxia inducible factor-1,HIF-1)表达,HIF-1 通过其下游的信号传导途径促进肿瘤细胞增殖、启动肿瘤血管新生、躲避细胞凋亡程序等,同时HIF-1反过来可直接促进肿瘤细胞糖酵解。
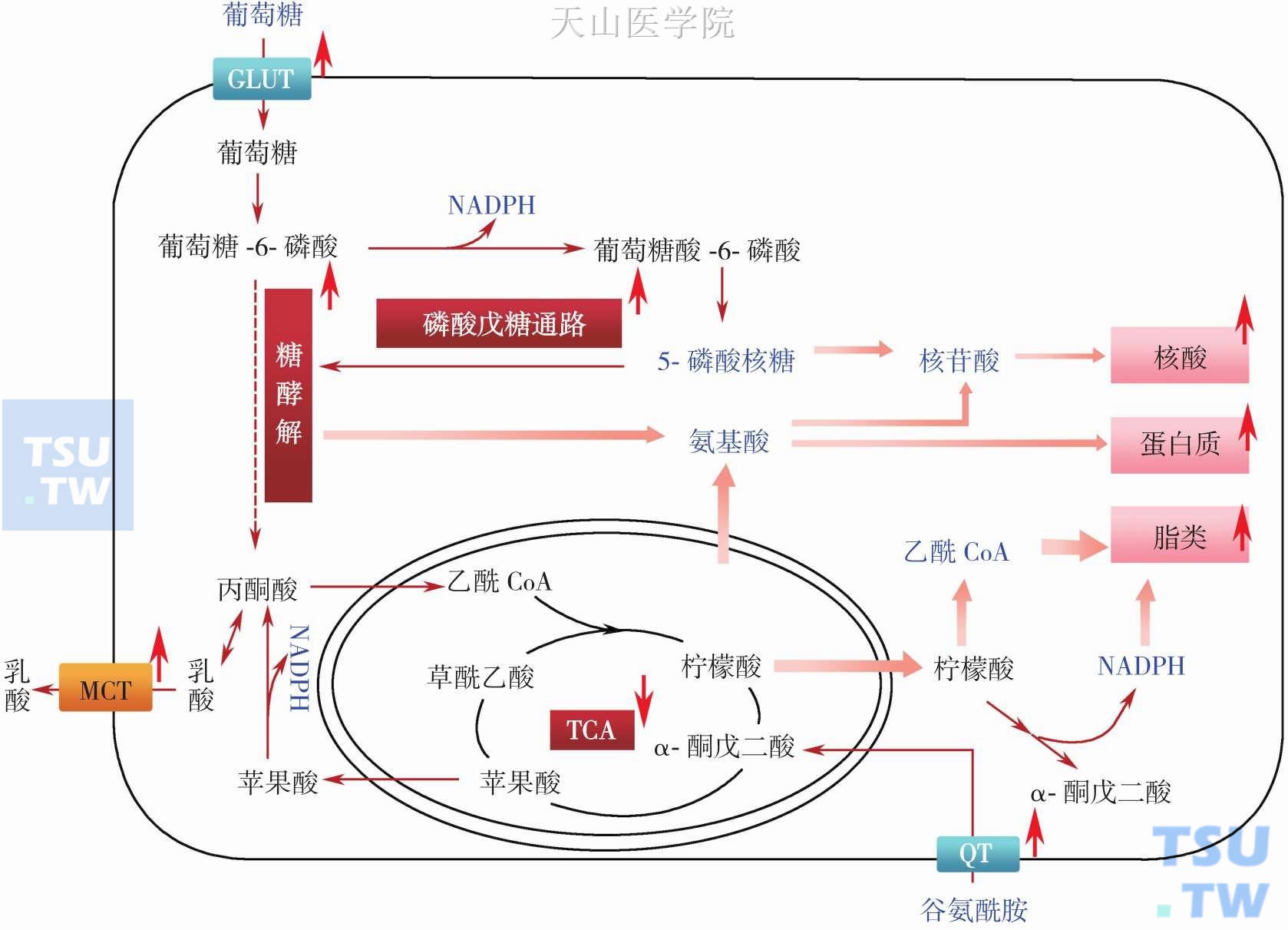
图3-2-3 肿瘤代谢重编程促进合成代谢
肿瘤Warburg效应的发生机制:目前关于肿瘤细胞的Warburg 效应发生机制并没有完全阐明。可能的机制:包括癌基因和抑癌基因异常(突变、缺失、增加和表观异常修饰),糖代谢酶和转运载体异常(突变、表达和同工酶),线粒体结构与功能异常,以及缺氧诱导因子1(HIF-1)等。
(1) 癌基因激活和抑癌基因失活是导致肿瘤发生Warburg效应的主要原因:调控细胞增殖的生长信号通路同样调控细胞的代谢。肿瘤细胞内癌基因激活和抑癌基因失活致生长信号通路异常激活从而导致重编程细胞代谢(图3-2-4):由癌基因产物(绿色表示)组成的主要生长信号通路有两条,而抑癌基因产物(红色表示)起抑制作用。当生长因子作用于膜受体激活受体酪氨酸激酶(RTK),后者可分别激活PI3K-Akt和Ras-Raf-ERK通路,两者最终可抑制TSC1-TSC2复合物形成,从而使信号通路的开关分子G蛋白Rheb处于GTP结合的活化状态(Rheb-GTP);Rheb-GTP激活mTORC1,后者通过不同机制(磷酸修饰转录和翻译相关蛋白促进相关代谢基因表达)介导肿瘤细胞代谢重编程。
磷脂酰肌醇-3-激酶(phosphoinositide 3-kinase,PI3K) 信号通路可被多种生存信号如细胞因子、生长因子、激素和癌蛋白Ras等激活。PI3K是一个异源二聚体(85kD调节亚基和110kD的催化亚基)蛋白。一旦G蛋白耦联受体或酪氨酸激酶受体被激活,85kD调节亚基通过SH2结构域与受体上磷酸化的酪氨酸结合并激活110kD的催化亚基,后者相继催化膜上肌醇磷脂的磷酸化,包括磷脂酰肌醇-4,5-双磷酸(PIP2)磷酸化生成具有第二信使的磷脂酰肌醇-3,4,5-三磷酸(PIP3)。PIP3可募集和激活3-磷酸肌醇依赖的蛋白激酶(PDK1),PDK1可激活蛋白激酶B(PKB/Akt),接着PKB/Akt激活下游靶蛋白产生相应的效应。肿瘤细胞中Akt/ PKB激活大多由于抑癌基因蛋白PTEN失活,因为PTEN可催化PIP3脱磷酸回复到PIP2,从而阻止PKB/Akt信号通路。人类肿瘤中大多有PTEN突变而失活,因此,肿瘤细胞中在没有生长因子受体激活的情况下也可激活Akt/PKB。
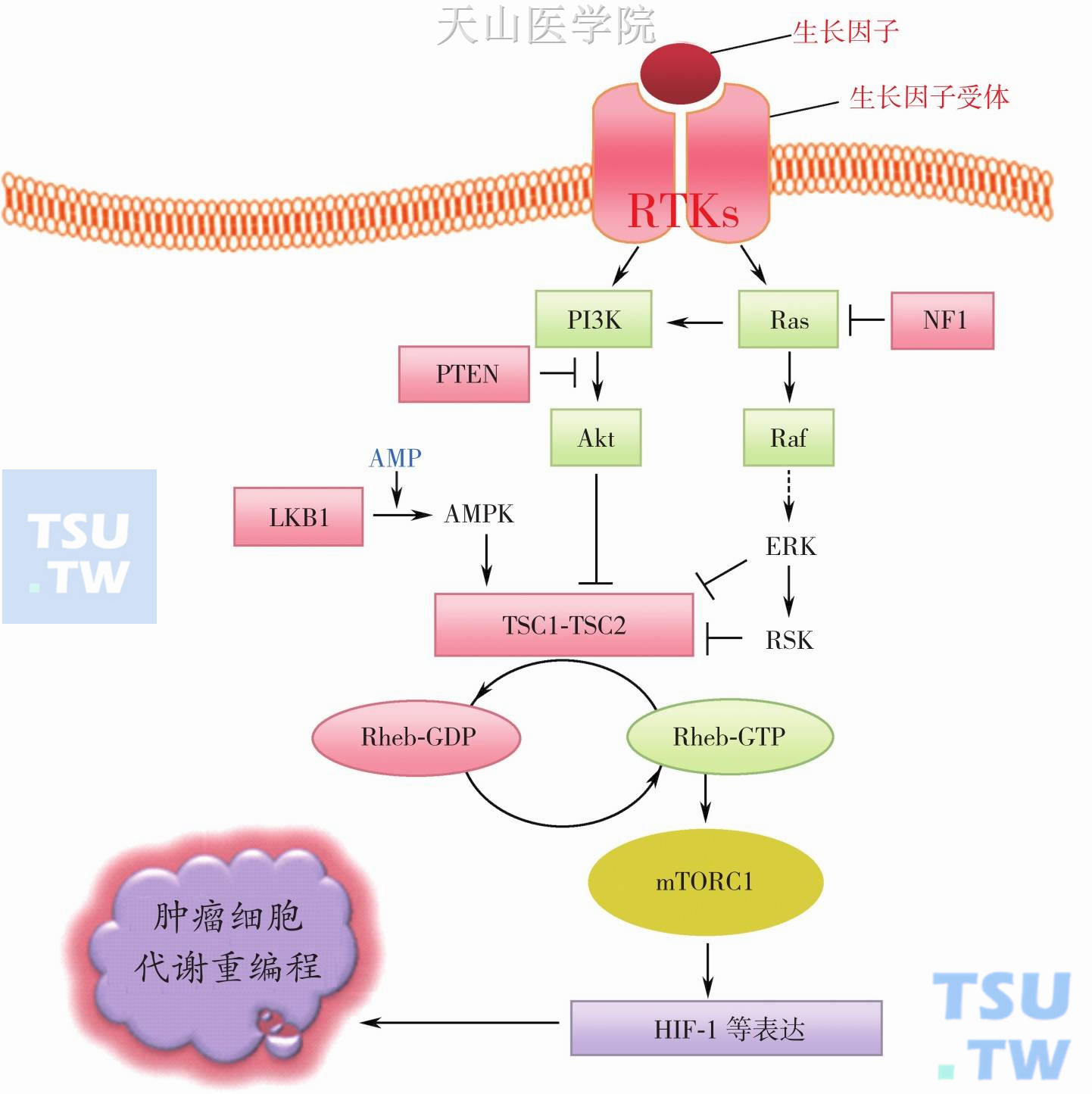
图3-2-4 癌基因激活和抑癌基因失活导致生长信号通路异常激活和代谢重编程
RTKs:受体酪氨酸激酶;PI3K:磷脂酰肌醇3激酶;Akt/PKB:蛋白激酶B;Ras:一种小分子GTP结合蛋白;Raf:一种丝/苏氨酸蛋白激酶;ERK(MAPK):细胞外信号调节激酶(丝裂原激活蛋白激酶);RSK:核糖体S6激酶;Rheb:脑中富含的Ras同源分子(GTP结合蛋白);mTORC1:哺乳动物雷帕霉素靶蛋白复合物1(丝/苏氨酸蛋白激酶);AMPK:AMP激活的蛋白激酶;TSC:结节状硬化症复合物;PTEN:磷酸酶和张力蛋白同源;LKB1:肝脏蛋白激酶B1(丝/苏氨酸激酶11,STK11);NF1:神经纤维瘤蛋白1
PI3K/Akt信号通路激活主要促进细胞生存和增殖,但也可促进葡萄糖摄入和糖酵解和磷酸戊糖通路酶活性(图3-2-5):PI3K/Akt信号通路促进葡萄糖转运载体(glucose transporters,GLUT)1和3表达并在肿瘤细胞膜上成簇分布;增加HKⅡ表达和活性,并通过PI3K/Akt/mTOR促进其与线粒体结合,Akt 还可显著调节糖酵解酶如6-磷酸果糖激酶-2表达,以及己糖激酶转位至线粒体外膜上等,这样促进葡萄糖磷酸化和更多葡萄糖进入细胞代谢;PI3K/Akt 信号通路还可增加其他糖酵解酶如6-磷酸果糖激酶-1(PFK1)表达等。最新研究发现异常激活的mTOR通过诱导产生胚胎型丙酮酸激酶M2(PKM2)等。此外,线粒体呼吸功能损伤(如mtDNA突变)也可激活PI3K/Akt信号通路。线粒体呼吸下降可失活PTEN,导致PI3K/Akt信号通路激活。当肿瘤细胞mtDNA丢失后也可激活PI3K/ Akt信号通路,同时伴有糖酵解,增强细胞侵袭和凋亡逃逸等。因此,除了生长因子直接激活和增强PI3K活性和PTEN突变和下调外,肿瘤细胞普遍存在的线粒体呼吸功能损伤也可增强PI3K/Akt信号通路。
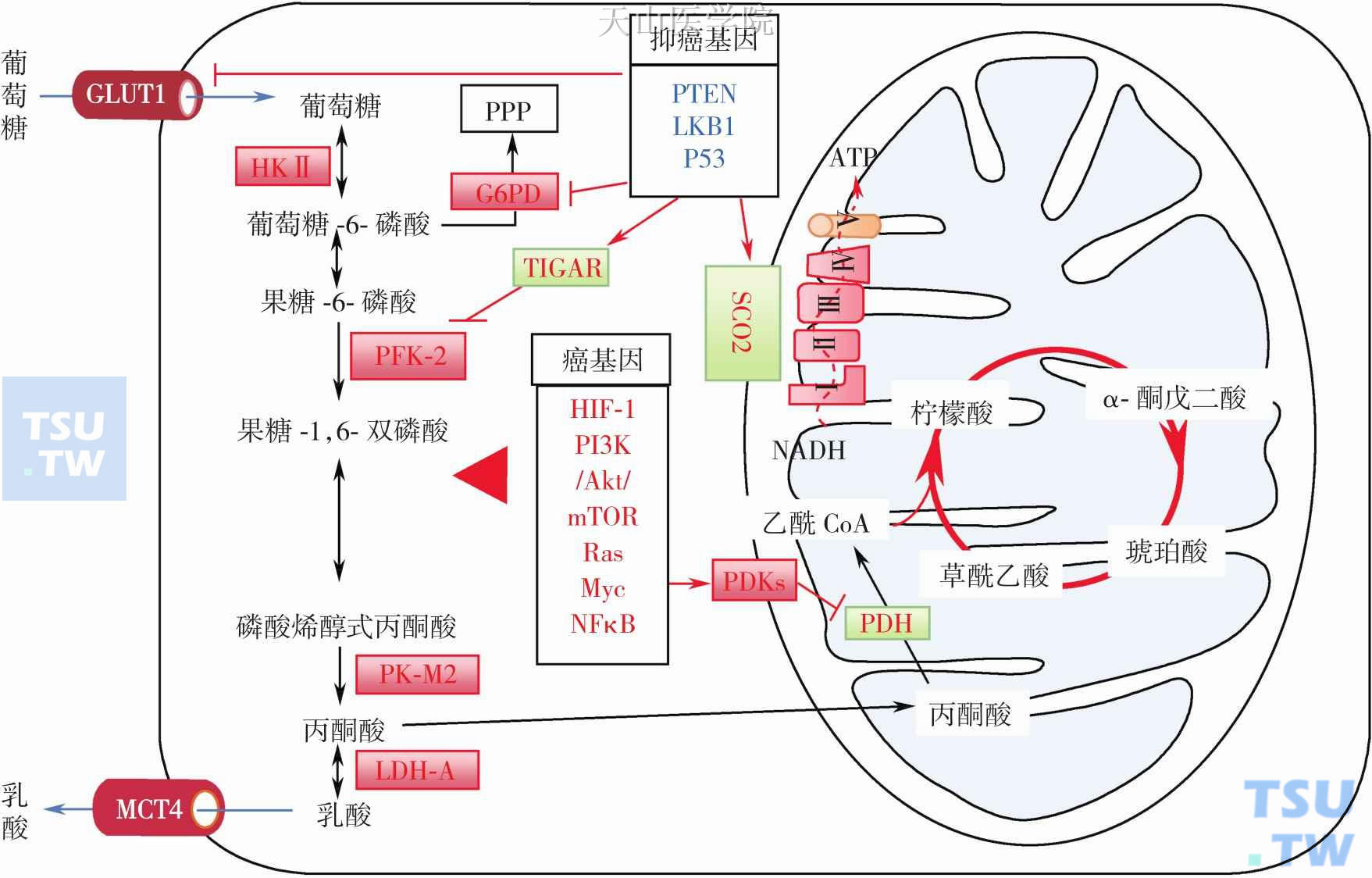
图3-2-5 癌基因激活和抑癌基因失活影响葡萄糖代谢酶和转运载体活性
除了参与上述生长信号通路发挥调节肿瘤代谢重编程的癌基因外,还有一些激活癌基因编码蛋白如Myc和Mondo A等直接调节相关代谢酶表达发挥作用。Myc是一个转录因子,在许多肿瘤细胞中高表达。Myc通过几个途径促进糖酵解:Myc可以直接与HKⅡ、aldolase、GAPDH、enolase,PK和LDHA基因启动子结合,诱导糖酵解酶表达;Myc激活可引起细胞内ROS升高,损伤mtDNA和线粒体呼吸功能,导致糖酵解增强。Mondo A类似Myc具有调节糖酵解的转录因子,Mondo A分子内含有螺旋-环-螺旋的亮氨酸拉链模体,与M1x结合形成异源二聚体。Mondo A-M1x复合物可在线粒体和细胞核之间迁移,可感受线粒体的能量产生状况,并将此信息传递至胞核激活相关靶基因表达。糖酵解的三个关键酶(HKⅡ、LDHA和PFK-2/ FBPase)的基因是直接受Mondo A-M1x复合物调节的靶基因。其他如Ras 和Src通过增加GLUT的表达增加糖摄取。H-ras 可以促进糖酵解,且降低氧消耗,而抑制H-ras 表达,可使HIF-1a 和糖酵解酶活性也降低,抑制膀胱癌U87 糖酵解并导致细胞死亡。癌蛋白Bcr-Abl促进Warburg 效应的作用能被Bcr-Abl的抑制剂Gleevec逆转,使糖代谢从糖酵解向线粒体氧化磷酸化转变。可见,肿瘤基因信号可能在能量调节中起着重要的作用,是造成Warburg效应的一个原因。
抑癌基因突变失活或缺失而失去负性调控生长信号通路和糖代谢稳态调节(图3-2-4和图3-2-5)。如P53可通过负调PI3K/Akt -mTOR信号通路来抑制糖酵解;P53还直接抑制GLUT 1和4,以及间接通过抑制NF-κB信号通路降低GLUT3表达来降低细胞摄取葡萄糖;p53可通过转录激活TIGAR (TP53-induced glycolysis and apoptosis regulator)表达来抑制糖酵解。TIGAR具有磷酸酶活性,能使果糖-2,6-二磷酸去磷酸化生成果糖-6-磷酸,降低细胞内果糖-2,6-二磷酸水平,而后者是磷酸果糖激酶-1 (PFK-1)的强力激活因子;P53通过激活磷酸葡萄糖变位酶 (phosphoglycerate mutase,PGM)的泛素化,促进PGM蛋白质降解而抑制糖酵解。p53还可调节帕金森氏病相关基因Parkin来抑制糖酵解。此外,最近研究发现P53调节PPP机制主要通过非转录方式直接影响PPP关键酶葡萄糖-6-磷酸脱氢酶 (G6PD) 活性,缺失P53 的MEF 和HCT116 细胞内G6PD活性明显增强,在U2OS细胞内RNA干扰下调P53表达后,G6PD活性是对照组的二倍,P53缺失的小鼠组织中G6PD活性也明显升高。通过一系列实验证实P53可以与G6PD单体结合而阻止形成G6PD二聚体的活性形式,从而降低细胞葡萄糖消耗、NADPH合成和大分子合成。正常组织细胞内野生型P53表达后能很好地限制PPP代谢流量,使得葡萄糖主要进入氧化分解途径。但是大部分肿瘤细胞中,P53往往发生突变或缺失,使得P53失去与G6PD结合和抑制能力,导致大量葡萄糖进入糖酵解和PPP代谢。其他抑癌基因包括PTEN和LKB1失活也将产生类似作用。
(2) 糖代谢酶和转运载体异常:如上述由于癌基因和抑制基因异常导致生长信号通路激活可引起葡萄糖转运载体和代谢酶表达异常,导致肿瘤细胞糖代谢异常。同时还有这些编码基因本身结构和功能异常,如突变酶或异常同工酶谱等。通常肿瘤细胞在选择同工酶时,优先表达古老型或胚胎型同工酶,这类同工酶具有多方面酶动力学优势:底物亲和力高、高催化活性、无产物抑制等。研究发现整个糖酵解和磷酸戊糖通路中很多关键酶表达和同工酶发生改变:如己糖激酶(hexokinase,HK)、磷酸果糖激酶(phosphofructokinase,PFK)、丙酮酸激酶(pyruvate kinase,PK)、乳酸脱氢酶(lactic dehydrogenase,LDH)和磷酸甘油醛脱氢酶(phosphoglyceraldehyde dehydrogenase,GAPDH),葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD)和转酮醇酶(transketolase,TKL)等(图3-2-5,图3-2-6)。
HK:是糖酵解的第一个限速酶。人类细胞有4种HK亚型,它们有不同的组织和细胞内分布,其中,HKⅡ与肿瘤相关性最大。正常情况下,HKⅡ仅在脂肪、肌肉和心肌组织中微量表达。Sebastian等发现很多肿瘤细胞系中都有HKⅡ过表达,在许多生长迅速的恶性肿瘤细胞中高表达。HKⅡ表达增多,从基因水平上而言,首先是其基因复制的增多;另一个重要的原因就是HKⅡ的启动子有广泛的信号转导级联激活途径(胰岛素、乏氧环境、佛波酯、突变型的p53和HIF-1α所激活)。除催化活性外,HKⅡ还可拮抗线粒体途径的细胞凋亡。肿瘤细胞倾向优先表达HKⅡ有代谢上的几个理由:HKⅡ较HKⅣ(葡萄糖激酶)有更高的底物亲和力,是后者的100倍,从代谢角度对肿瘤细胞相当有利;HKⅡ与HKⅢ和HKⅣ结构有些不同,其N端含有一个疏水结构域,使之能够与线粒体mPTP复合物的VDAC结合,HKⅡ与VDAC的结合赋予了酶促动力学上的多种优势:使HKⅡ与ATP结合亲和力提高约5倍,优先获得线粒体释出的ATP,并且其酶促反应不受葡萄糖-6-磷酸产物的负反馈抑制;尽管HKⅠ和HKⅡ都含有两个相当于葡萄糖激酶结构域,但只要HKⅡ的两个结构域都具有催化活性。基于上述理由,需要高酵解速率的肿瘤细胞选择HKⅡ。
PFK:催化果糖-6-磷酸生成果糖-1,6-双磷酸(F1,6BP),是控制酵解速度最主要的限速酶。而磷酸果糖激酶-2(PFK-2)催化果糖-6-磷酸生成果糖-2,6-双磷酸(fructose 2,6-bisphosphate,F2,6BP),果糖-2,6-双磷酸可以别构激活PFK-1而促进糖酵解,抑制糖异生作用。细胞内,果糖-2,6-双磷酸浓度依赖于PFK- 2/果糖- 2,6-双磷酸酶(FBPase)的相对活性。PFK-2/ FBPase是双功能糖酵解酶,有2个分开的催化中心,分别催化6-磷酸果糖2位磷酸化和果糖2,6-双磷酸2位去磷酸。PFK-2通过影响2,6-双磷酸果糖水平实现对糖酵解通路的调节。在许多肿瘤细胞中,果糖2,6-双磷酸比正常的组织细胞的水平高。将编码PFK-2的基因导入肿瘤细胞,可增强细胞糖酵解活性。
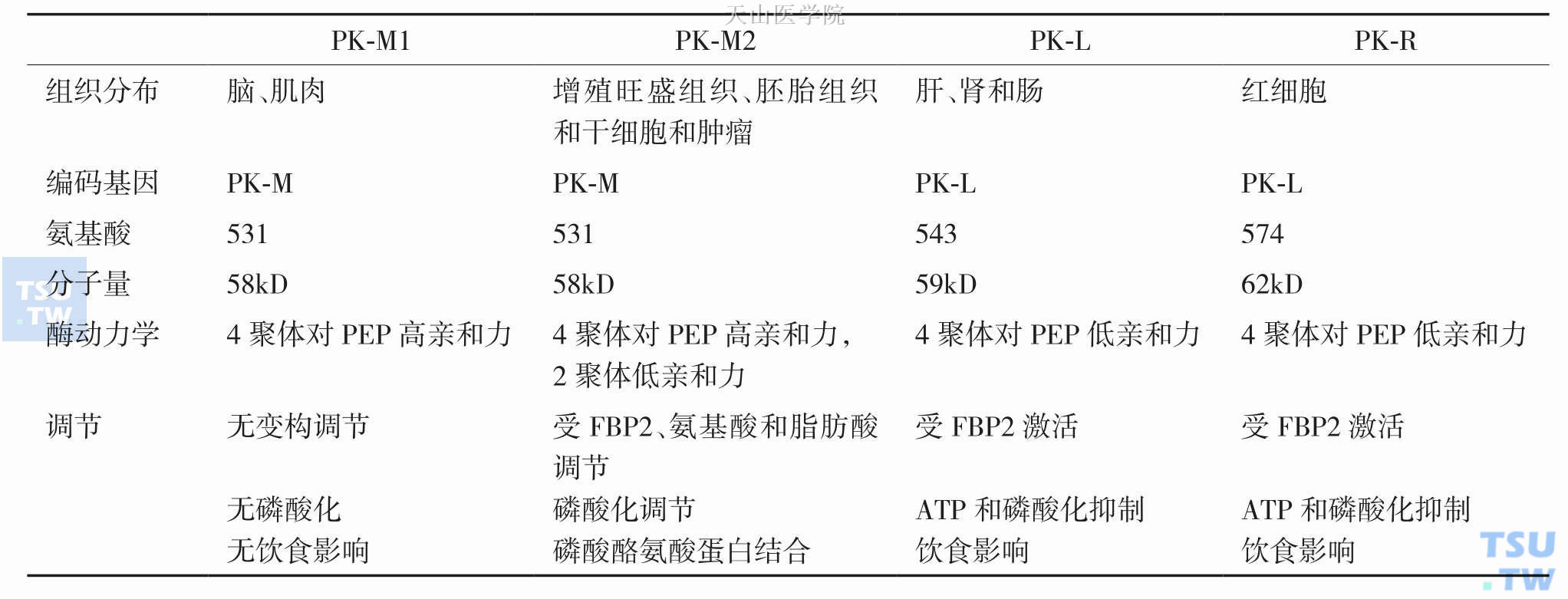
PK:有四种同工酶,分别是PK-L、-R、-M1 和-M2,它们的动力学特性、分布和作用是不同的(表3-2-3)。PK-M2为胚胎型丙酮酸激酶,它是糖酵解途径的关键酶,一些研究表明肿瘤细胞的恶性程度与PKM2的过度表达密切相关。有研究认为PK-M2可能在肿瘤细胞转换为有氧酵解中起一个开关的作用,ChristofkHeather R等用RNA干扰下调PK-M2或用PK-M1置换后逆转肿瘤细胞Warburg效应并降低裸鼠成瘤能力。PK-M2存在四聚体和二聚体结构类型,通过高活性的四聚体和无活性的二聚体之间转换可调节肿瘤细胞的糖酵解流量。四聚体的PK-M2与糖酵解其他酶结合在一起的高活性状态有利于糖酵解代谢;而二聚体的PK-M2游离的失活状态存在于胞质中,此时有利于为细胞生物合成提供代谢中间物(图3-2-6)。这两者可进行周期性转换波动以满足肿瘤细胞能量和合成代谢的需求。在肿瘤细胞中调节PK两种构象转换主要受糖酵解中间物FBP和磷酸化修饰等影响。当细胞内为高浓度失活的二聚体PK-M2时,糖酵解中间物FBP浓度升高,当FBP达到一定浓度时,可变构激活PK-M2,使二聚体PK-M2重新结合形成高活性的四聚体,导致糖酵解加快,转而又使FBP浓度下降;当FBP浓度下降到一定水平时又促使PK-M2解聚为二聚体,糖酵解中间物增加而促进细胞合成代谢。PK-M2还受磷酸化调节,2008年Christofk Heather R等用蛋白质组技术筛选出一个能特异性与磷酸酪氨酸蛋白结合的蛋白质,鉴定证实是PK的一种剪切亚型PK-M2。当磷酸酪氨酸蛋白替代FBP结合变构位置后,PK-M2活性下降。这提示肿瘤细胞受生长因子刺激增殖时,磷酸化信号通路可下调PK-M2活性使糖酵解中间物转向用于细胞合成代谢,这对肿瘤细胞快速生长十分关键。2014年Liangqian Huang等发现了PK-M2调节分子热休克蛋白40(HSP40),HSP40-PK-M2结合后通过HSC70降低PK-M2稳定性,从而影响糖酵解和线粒体呼吸。
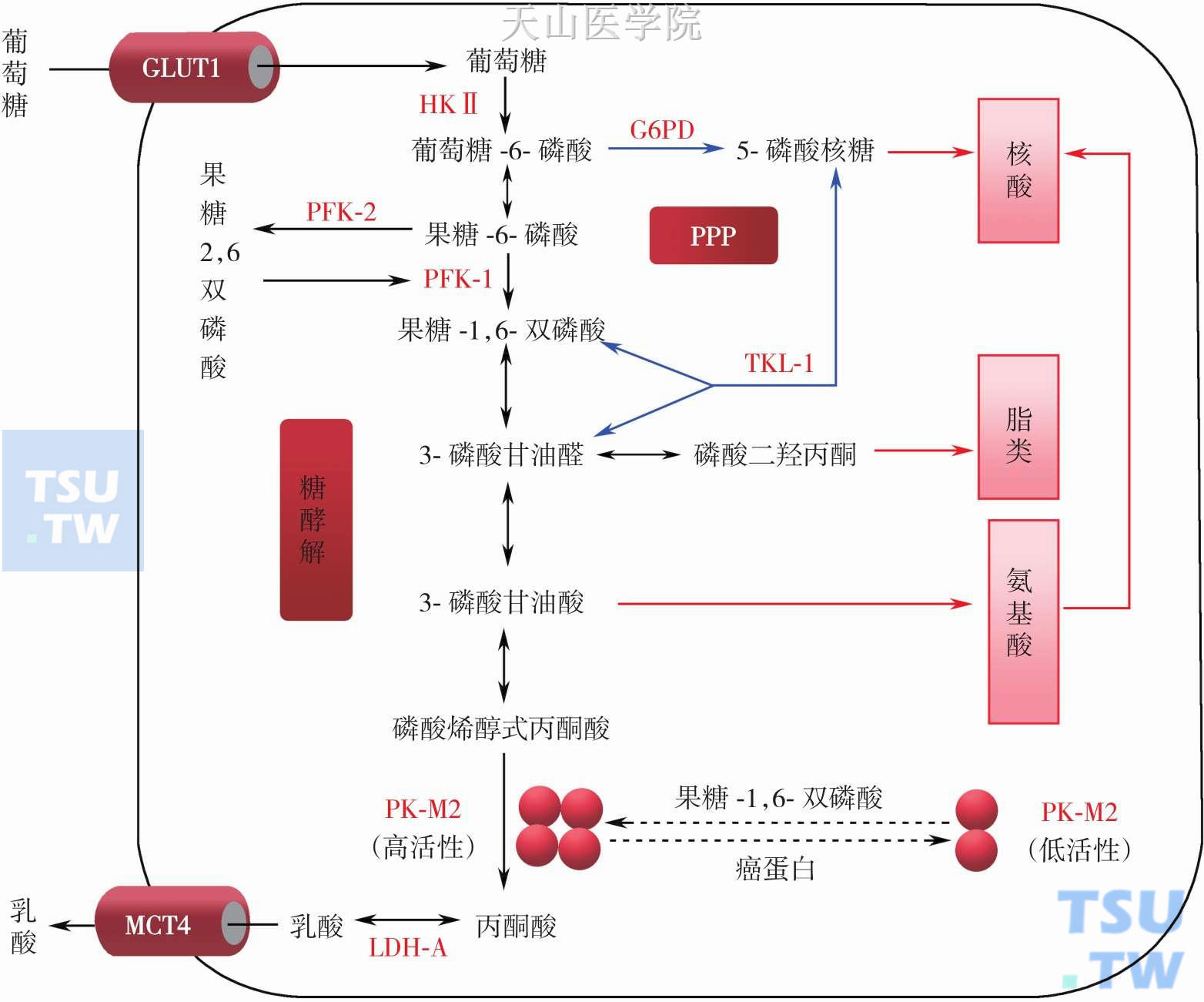
图3-2-6 肿瘤细胞糖代谢酶和载体异常同工酶和表达
GLUT 1,3:葡萄糖转运载体;HKⅡ:己糖激酶Ⅱ;PFK-1L和P型:磷酸果糖激酶-1;PFK- 2:磷酸果糖激酶-2;PK-M2:胚胎型丙酮酸激酶-M2;LDHA:乳酸脱氢酶A;G6PD:6-磷酸葡萄糖脱氢酶;TKL-1:转酮醇酶-1;MCT4:单羧酸转运载体4
表3-2-3 丙酮酸激酶同工酶分子和动力学特性
(3) 缺氧诱导因子(HIF)是细胞代谢重编程的重要癌性分子:肿瘤最主要的特征是细胞失控生长,肿瘤实体增大,由于供血跟不上瘤体组织生长速度,造成肿瘤内缺氧微环境的形成。在快速生长的肿瘤细胞中,供氧量不能满足线粒体产生ATP的需求,肿瘤细胞继而通过上调糖酵解来补偿氧化磷酸化产能的不足,逃避缺氧导致的死亡。缺氧诱导因子(hypoxia-inducible factor,HIF)在其中发挥了重要作用。1992年Semenza和Wang首先在缺氧诱导的细胞核抽提物中发现的。HIF-1是一种异源二聚体,由120kD的HIF-1α和91~94kD的HIF-1β两个亚单位组成。HIF-1β亚基又称芳香烃受体核转运蛋白(aryl hydrocarbon receptor nuclear translocator,ARNT),基因定位于人的1号染色体q21区,在细胞内稳定表达并起结构性作用,HIF-1α基因定位于人的14号染色体q21~24区,受缺氧信号的调控,是HIF-1的活性亚基。每个亚单位的氨基端均含有碱性的螺旋-环-螺旋(bHLH)结构域和Per/Amt/Sim(PAS)结构域,是其形成异源二聚体并与DNA结合所必需的结构。作为活性亚基的HIF-1α,由826个氨基酸构成,其两个末端是感受缺氧信号的活性调控区域,C末端有一个富含脯氨酸-丝氨酸-苏氨酸(Pro/Ser/Thr)的氧依赖降解结构域(oxygen dependent degradation domain,ODDD)和反式激活结构域(transactivation domain,TAD),即TADC,N末端含有TADN,这些结构域都是缺氧诱导蛋白稳定、核定位和转录激活的调节域,其中TADC发挥精细调节作用,TADN为激活转录所必需,可见HIF-1α亚基受缺氧调控并调节HIF-1的活性。关于HIF-1β,除了结构性组成作用外,其还可能与HIF-1在核内的稳定性及二聚化后的构象转变有关。有研究证明在ARNT缺陷的细胞不能诱导HIF-1的活性,HIF-1α亚基必须与HIF-1β亚基聚合形成异二聚体,才能发挥转录因子的作用。HIF-1可与一系列靶基因,包括红细胞生成素、血管生长因子(VEGF)、葡萄糖转运载体-1(GLUT-1)和糖酵解关键酶的低氧反应元件(hypoxia-responsive elements,HREs)结合激活基因转录。
HIF-1在正常供氧条件下有一定量表达,但很快被细胞内氧依赖性泛素蛋白酶降解途径所降解,只有在缺氧条件下HIF-1才可稳定表达。HIF-1活性取决于HIF-1α的稳定性,HIF-1α的稳定性受多种因素调节。首先是HIF-1α分子结构中有两个脯氨酸羟化位点(P402和P564),其羟化是依赖氧浓度的,并在3种脯氨酸羟化酶(PDHs)之一催化完成。羟化后的HIF-1α易于被von Hippel-Lindau (VHL)-Elongins复合物识别和结合,并介导泛素化,泛素化的HIF-1α随后被蛋白酶体复合物降解。VHL作为肿瘤抑制因子,编码泛素化连接酶的一个亚基,在正常氧浓度下,通过介导HIF-1α降解而抑制肿瘤发生(图3-2-7)。因此,在低氧调节下,HIF-1α羟化减少而其稳定性提高,导致HIF-1浓度增高而转录激活一系列靶基因表达,造成肿瘤细胞代谢表型。此外,当细胞氧浓度降低时,HIF-1α的转录和翻译呈指数增加,故称其为氧敏感亚基。HIF-1β是组成性表达的亚基,在常氧和缺氧状态下均可表达,它与HIF-1α结合成二聚体形成有活性的HIF-1,抵抗蛋白水解酶的降解。缺氧环境可诱导HIF-1α的表达,继而上调肿瘤细胞糖酵解途径。三羧酸循环累积底物,如琥珀酸、延胡索酸和2-羟戊二酸(突变异柠檬酸脱氢酶催化α-酮戊二酸还原产物)可竞争性抑制依赖α-酮戊二酸的HIF-1α脯氨酸羟化酶,导致HIF-1α的稳定和激活。因此这类堆积的代谢物称为致癌性代谢物(oncometobolite)。一般认为HIF-1仅在低氧情况下才稳定和有功能,而Warburg效应是在正常氧情况下糖酵解增强,因此肿瘤细胞在有氧情况下HIF-1很可能出现了异常的活化状态。某些肿瘤在充氧环境下也观察到高水平的HIF-1α表达,这表明除了低氧,其他因素如激素和生长因子等也可诱导HIF-1的稳定表达。一些研究表明PI3K/Akt-mTOR信号通路是上调HIF-1α的关键信号分子,有研究发现缺失TSC肿瘤抑制因子细胞只激活mTOR就可驱动HIF依赖的糖酵解和血管生长相关靶基因的表达。最新研究发现PTEN诱导性激酶-1(PTEN-induced kinase-1,PINK1)可降低HIF-1稳定性,PINK1的这个作用是通过线粒体产生ROS实现的。
HIF-1是具有转录活性的核蛋白,具有相当广泛的靶基因谱,其中包括与缺氧适应、炎症发展及肿瘤生长等相关的近100种靶基因。当其与靶基因结合后,通过转录和转录后调控使机体产生一系列反应,有些反应尽管带有适应代偿性质,但也常给机体带来病理性损害,如低氧性肺动脉高压,肿瘤加速生长等。HIF-1与肿瘤Warburg效应密切相关,可促进葡萄糖摄入和糖酵解,以及抑制线粒体呼吸等相关基因的表达(表3-2-4),HIF-1还可通过调节血管生成靶基因表达促进肿瘤细胞增殖等。因此,许多研究者认为,HIF-1是肿瘤细胞糖酵解增强的原因之一。近期研究表明,HIF-1α还可通过转录活化丙酮酸脱氢酶激酶(PDK1)来抑制线粒体有氧呼吸。PDK1可以使丙酮酸脱氢酶(PDH)失活,抑制三羧酸循环和氧化磷酸化,致使细胞糖代谢由线粒体氧化磷酸化方式向糖酵解转变。有研究发现HIF-1可与表达失调的转录因子Myc协同方式诱导HKⅡ和PDK1促进Warburg效应,还可协同诱导VEGF的表达。
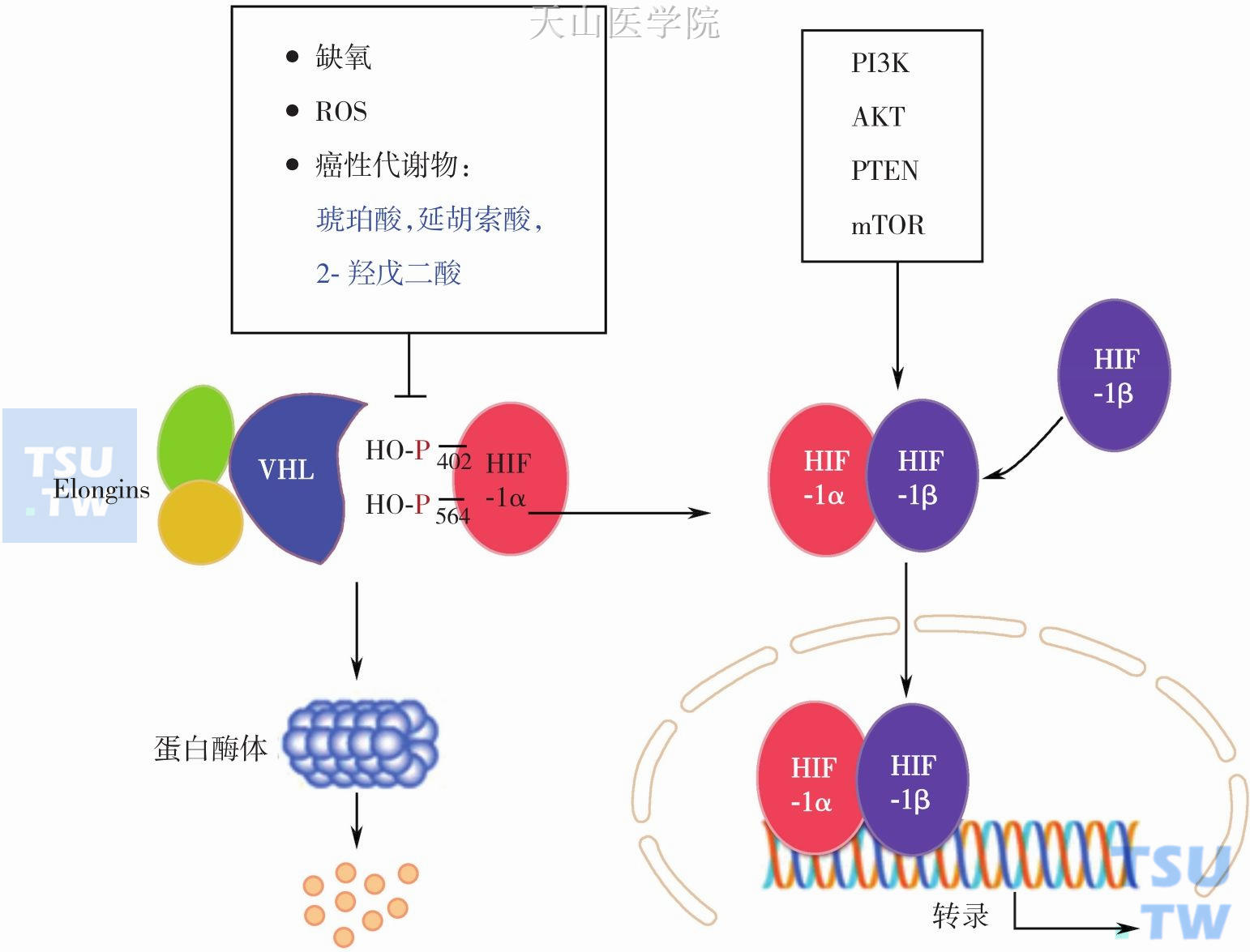
图3-2-7 HIF-1表达和稳定性调节
表3-2-4 HIF-1调节糖代谢靶分子

注:GLUT,葡萄糖转运载体;HK,己糖激酶;PGI,磷酸葡萄糖异构酶;PFK,磷酸果糖激酶;GAPDH,3-磷酸甘油醛脱氢酶;PGK,磷酸甘油酸激酶;PGM,磷酸甘油酸变位酶;TPI,磷酸丙酮异构酶;PK,丙酮酸激酶;LDHA,乳酸脱氢酶A;PFK-2/ FBPase,6-磷酸果糖激酶-2/果糖-2,6-双磷酸酶;MCT,单羧酸转运载体;PDK,丙酮酸脱氢酶激酶;MXI,max interactor,Max相互作用因子;COX4-2,细胞色素氧化酶亚型2
(缪明永)
